This site uses cookies to provide you with a more responsive and personalized service. By clicking "Accept," you agree to the use of cookies on this site. Please read our Cookie Policy and Privacy Policy for more information on the use of cookies on this website.
GSDIa is a rare metabolic disorder that leads to impaired glucose production1
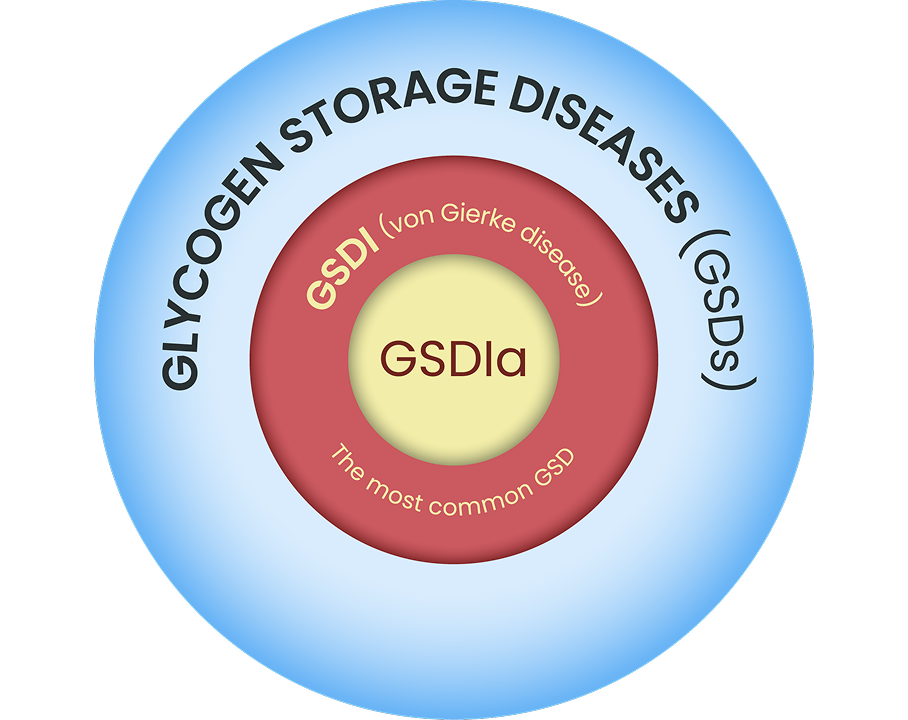
Image illustrative of disease types within the larger GSD hierarchy. Not to scale based on disease prevalence.
Glycogen storage disease type Ia (GSDIa, a von Gierke disease) is a metabolic disorder caused by a deficiency in the enzyme glucose-6-phosphatase (G6Pase) due to pathogenic variants of the G6PC (G6PC1) gene.1 This deficiency leads to an inability to release glucose from stored glycogen or generate glucose endogenously, especially during periods of fasting or high energy demands, leaving patients at risk of severe, life-threatening hypoglycemia.1,2
Pathophysiology of GSDIa
Unaffected Glucose Production
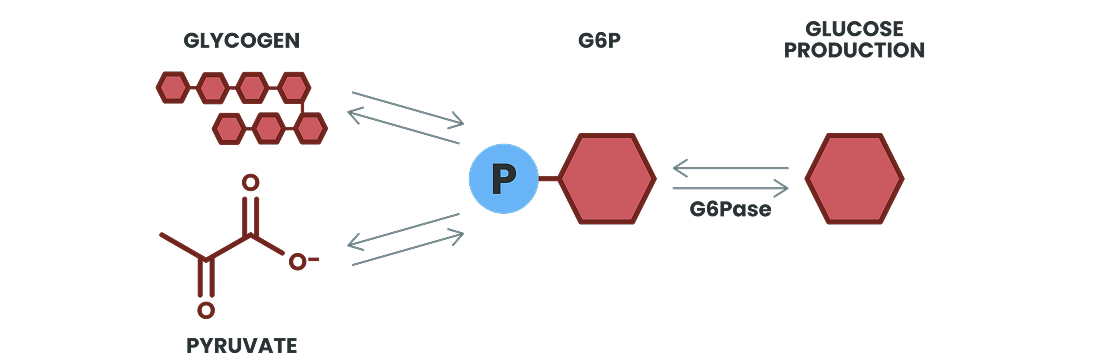
Glucose is produced endogenously to maintain normal blood glucose levels and is increasingly used during periods of fasting or high energy demands1,2
G6Pase is a critical enzyme in the production of glucose, as it catalyzes the conversion of G6P to
glucose during the final step of both glycogenolysis and gluconeogenesis1
G6Pase Deficiency in GSDIa
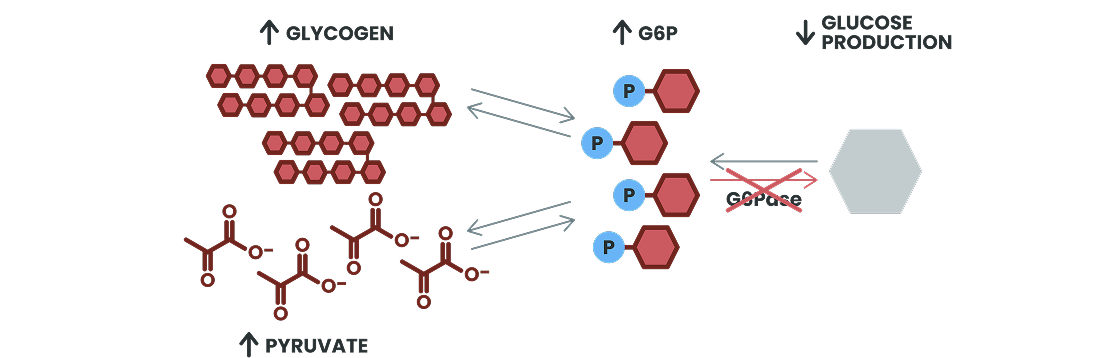
Without functional G6Pase, impaired glycogenolysis and gluconeogenesis result in
an inability to release glucose, causing potentially life-threatening episodes of hypoglycemia,
especially during periods of fasting or high energy demands1,2
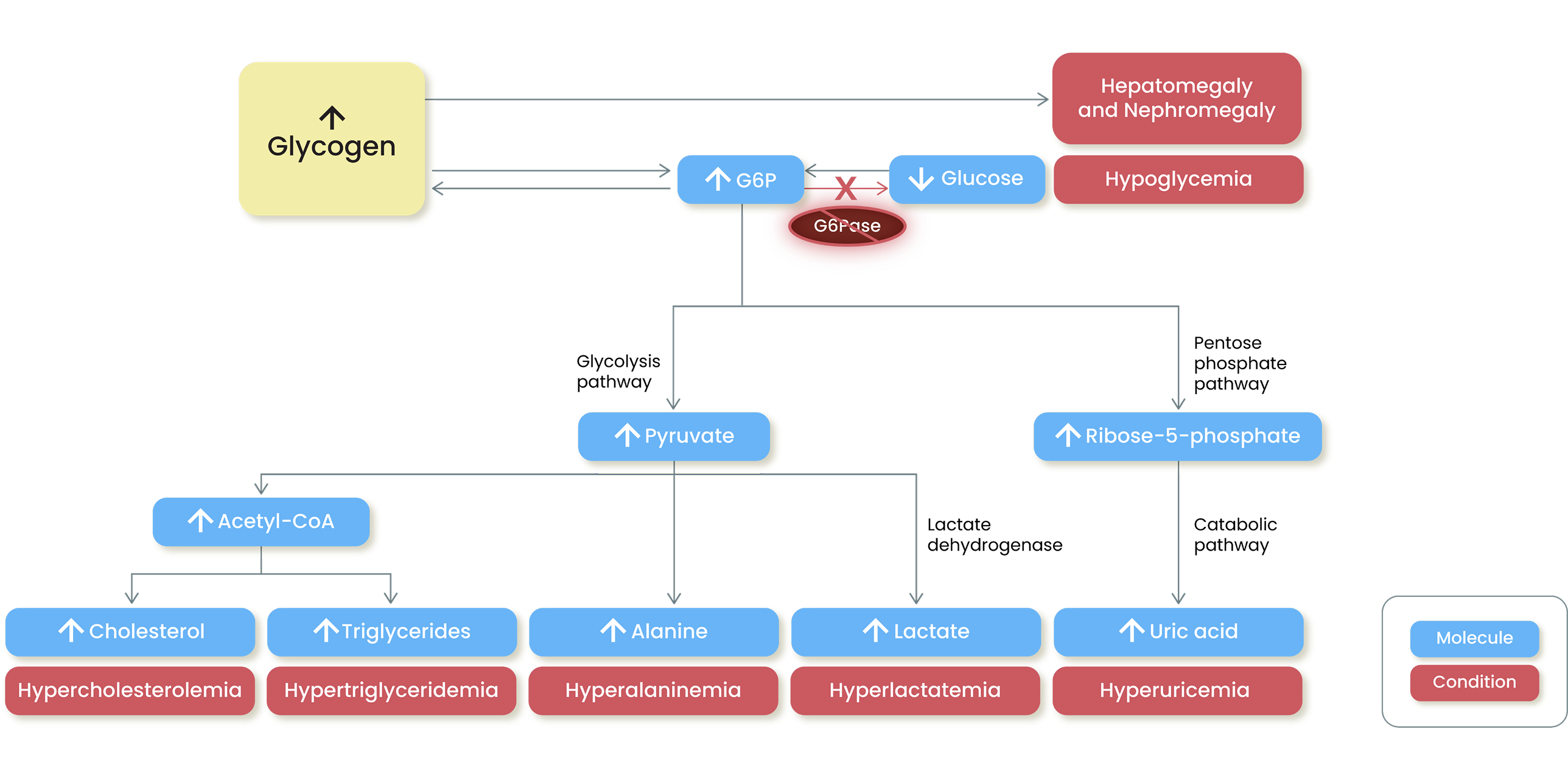
Excess glycogen builds up in the liver and kidneys, and accumulated G6P is shunted to alternative
metabolic pathways, resulting in multisystemic complications1
Image illustrative of disease types within the larger GSD hierarchy. Not to scale based on disease prevalence.


Abbreviations: G6P, glucose-6-phosphate; G6Pase, glucose-6-phosphatase; GSDIa, glycogen
storage disease type Ia; US, United States.
References: 1. Derks TGJ, Rodriguez-Buritica DF, Ahmad A, et al.
Glycogen storage disease type Ia: current management options, burden and unmet need. Nutrients.
2021;13(11):3828. 2. Kanungo S, Wells K, Tribett T, El-Gharbawy A.
Glycogen metabolism and glycogen storage disorders. Ann Transl Med. 2018;6(24):474. 3. National Organization for Rare Disorders. Glycogen storage disease
type I. Accessed July 1, 2025. https://rarediseases.org/rare-diseases/glycogen-storage-disease-type-i/