This site uses cookies to provide you with a more responsive and personalized service. By clicking "Accept," you agree to the use of cookies on this site. Please read our Cookie Policy and Privacy Policy for more information on the use of cookies on this website.
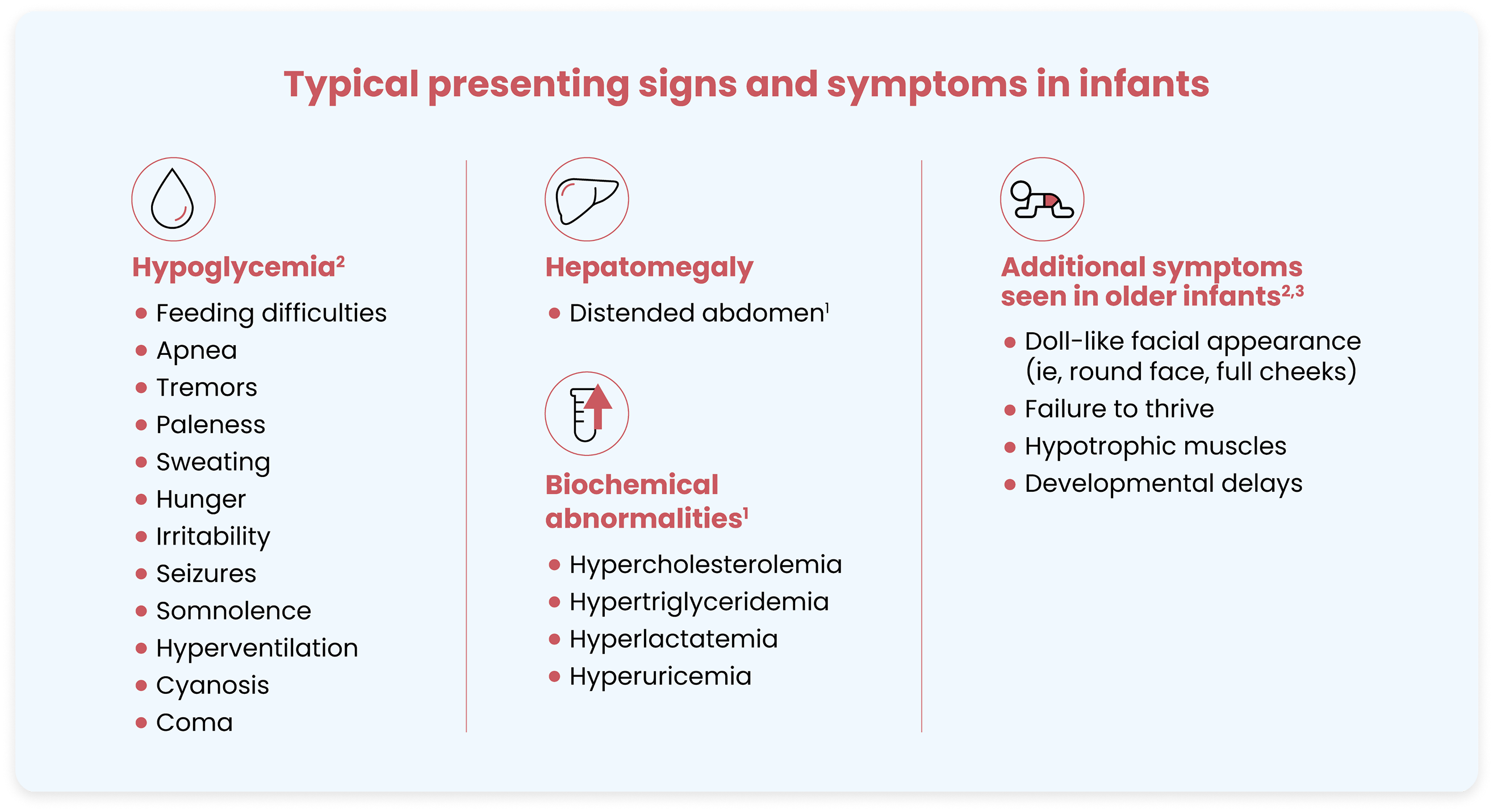
Infants with GSDIa present with fasting hypoglycemia, hepatomegaly, and biochemical and clinical abnormalities1
During the first few months of life, infants with GSDIa often remain asymptomatic, as frequent feedings provide adequate glucose to prevent hypoglycemia.1 Symptoms of GSDIa typically start to emerge between 3 to 6 months of age, as the interval between feedings increases.1
Genetic testing typically confirms a GSDIa diagnosis1
Abbreviations: G6Pase, glucose-6-phosphatase; GSDIa, glycogen storage disease type
Ia.
References: 1. Kishnani PS, Austin SL, Abdenur JE, et al. Diagnosis
and management of glycogen storage disease type I: a practice guideline of the American College of Medical
Genetics and Genomics. Genet Med. 2014;16(11):e1. 2. Derks
TGJ, Rodriguez-Buritica DF, Ahmad A, et al. Glycogen storage disease type Ia: current management options,
burden and unmet need. Nutrients. 2021;13(11):3828. 3. Özen
H. Glycogen storage diseases: new perspectives. World J Gastroenterol. 2007;13(18):2541-2553. 4. HRSA. Recommended uniform screening panel. Accessed August 1, 2025.
hrsa.gov/advisory-committees/heritable-disorders/rusp
5. Bali DS, El-Gharbawy A, Austin S, et al. Glycogen storage
disease type I. 2006. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews® [Internet]. Seattle
(WA): University of Washington, Seattle; 1993-2024. Accessed June 27, 2025. https://www.ncbi.nlm.nih.gov/books/NBK1312/